I’m pursuing a PhD in the field of structural biology. I’ve posted about it here before, (that thread is due for an update) but my university recently took possession of a large expensive microscope that is used to image small molecules like proteins, DNA, RNA, as well as larger biological assemblies such as viruses and cells. I’m currently responsible for the day-to-day operation of this microscope, as well as the computing infrastructure surrounding it, which is quite extensive.
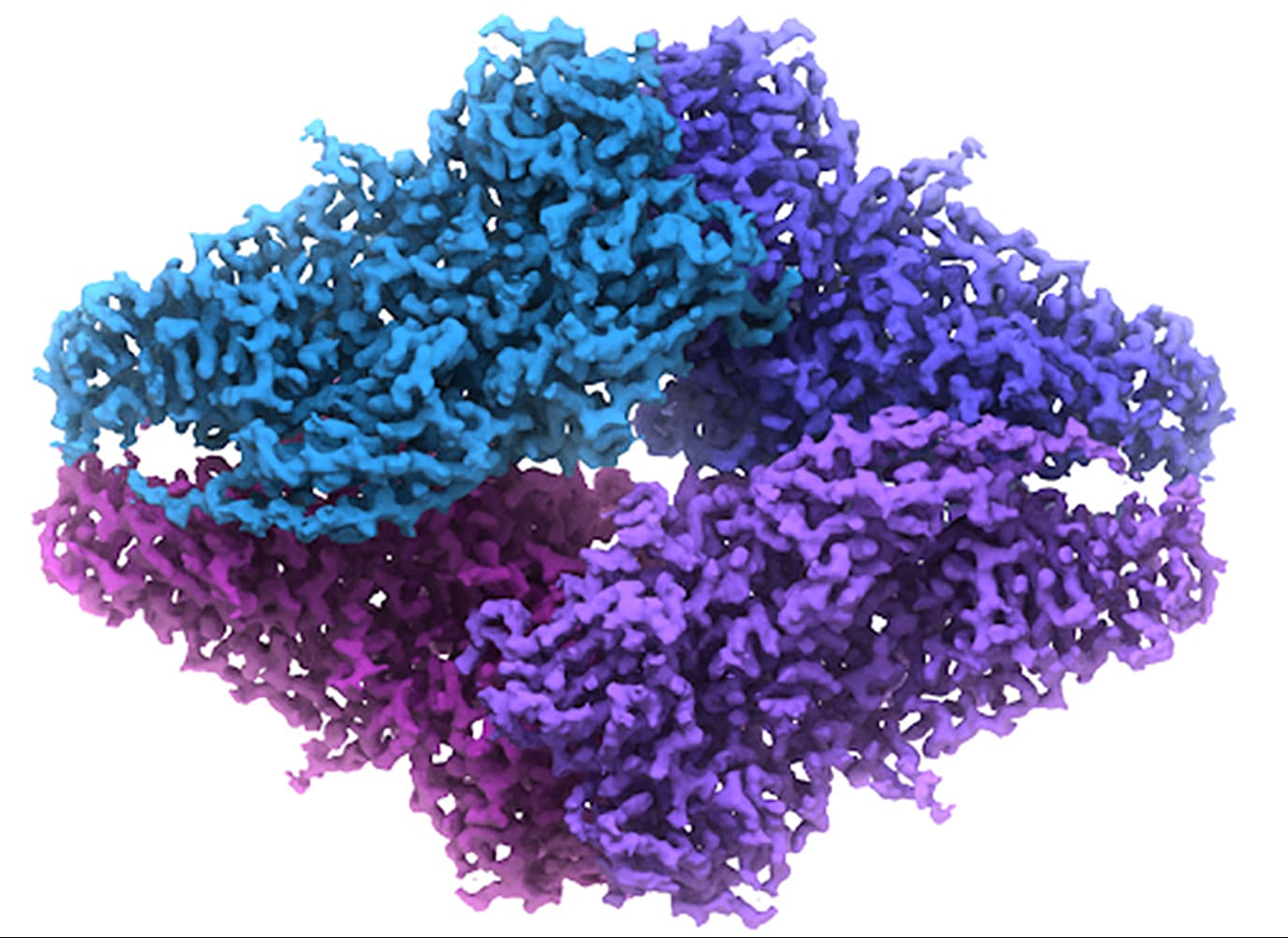
Experimental data in this field generally consists of a file that maps electron density to Cartesian coordinates at some specific pixel spacing, where the pixel spacing is representative of some distance in Angstroms or nanometers. It is, to my knowledge, always cubic. So at the end of the process of collecting and refining data of a protein, I might end up with what we call a “map” or “volume” that is essentially a 3D array of some size (say, 100x100x100) at some specific pixel spacing (say, 2.0 angstrom). That would mean I have a cube that is 200 cubic angstroms, and any position on a lattice in that cube has a corresponding value that represents the density of a pixel (or voxel) there.
Here is a 2D projection of what that looks like:
Then, if we know the amino-acid sequence of that protein, we can “build” it into that density, using rules we know about the shapes, sizes, bonds, angles, etc, of amino acids and peptides. The higher resolution the 3D map, the more confident we can be about the atomic model we build into it, and the more fine details we can model.
Here is an example of a protein atomic model being built into a 3D density map:
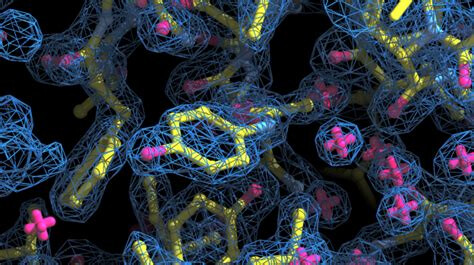
In this image, the map is the blue mesh, and the atoms and bonds are represented by the red (oxygen) and yellow (carbon).
In the old days of structural biology, the data coming from traditional methods was generally lower resolution. This means that to see a certain level of details that are only visible at higher resolutions, various techniques had to be employed to existing data to squeeze the maximum amount of information out of it. Some of these strategies were:
- Averaging data that had symmetry to improve the signal to noise. If you have data that has, say, 4 fold symmetry, you can expect real density to self-reinforce while noise will self-cancel (unless it’s systemic noise, but there are other strategies for dealing with systemic noise).
- Solvent flattening (fitting peaks to solvent in fourier space to help “sharpen” the map and distinguish between protein and non-protein density which allows for a less ambiguous fit)
- Masking and thresholding, which again rely on the fact that you can expect ordered regions to be your molecule of interest, and less-ordered regions to have lower density thresholds.
- High and low pass fourier filtering, which excludes wavelengths of specific frequencies to try and remove noise
…as well as a bunch more.
As the field of x-ray crystallography grew with the development of Synchotron beamlines that allowed for higher-resolution data collection, and the invention of CMOS image sensors in cryo-EM which also allowed for the collection of higher-resolution data, these strategies became less frequently used. As such, many of the people that developed and maintained the software for these techniques either moved on to other jobs, or retired and haven’t been replaced. In particular, the biggest loss was the Uppsala Software Factory software, which was a collection of tools for working with and manipulating 3D volume data to do all sorts of things with it. The tools hadn’t been updated since the early 2000’s, and last year, the main website, including all the documentation, went down.
This would be OK, because with high resolution data, they aren’t needed anymore, except that we have another exciting development in the field of structural biology that has actually made them quite useful: tomography.
While traditional structural data comes from imaging and classifying hundreds of thousands or millions of highly pure, homogenous particles over the course of weeks, and constructing that into one final model, tomography is a technique that lets us look at a single particle, but from multiple angles, and then combine those angles in fourier space to construct a 3D volume. However, because we are exposure limited (send too many 200 kV electrons at the same particle and it will degrade), the quality of this data tends to be of lower resolution.
One thing we can do, however, is extract particles from the 3D volume, and average them either together with other particles, or “locally”, to find symmetry on themselves and take advantage of that to increase our resolution.
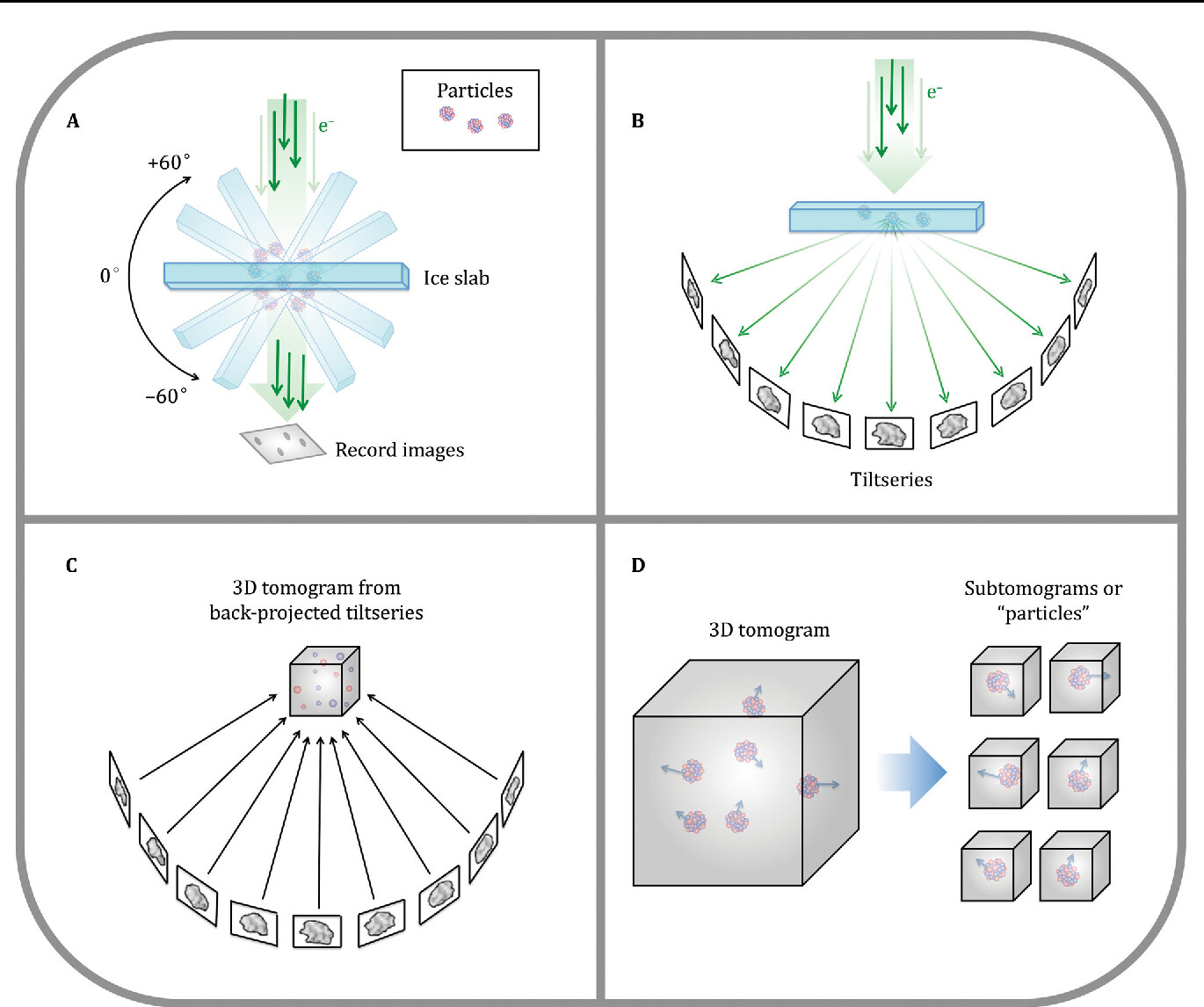
As I’ve been working with more and more low-resolution tomographic data, I’ve been lamenting the fact that the traditional tools for squeezing information out of low-resolution data, and manipulating and working with 3D volumes.
While the binaries themselves, and the source code, are present on GitHub (GitHub - martynwinn/Uppsala-Software-Factory: These are the famous Uppsala Software Factory programs, rescued into GitHub.), I’ve been unable to re-compile them as I know nothing about Fortran, and also, they are having a hard time with modern file formats for this type of data. I’ve written a couple of tools and scripts to make modern file formats compatible, but as what I’m trying to do is becoming more complex, that solution is failing rapidly.
My goal, then, is to begin to re-write/re-implement these tools in Python or another more-modern language, and to eventually release a full package capable of doing the bulk of the operations that these tools used to do. I’m terrible at math, and I’m terrible at programming, so I am going to recognize right now that this is a huge undertaking that will likely take me years, if it ever does succeed.
Therefore, for my Devember project, my goal is going to be more simple:
-
I want to develop a tool that will re-orient a 3D volume to be centered on its origin. Most modern software outputs these volumes so that their origin is 0,0,0 (the bottom left). I want to develop a tool that will move that origin to be the center of the volume. So that if you have a 100x100,100 volume, the origin is 50,50,50. This is because for averaging, it is much easier to assume that the origin is the center of the volume, and then to average symmetrically around that origin.
-
I want to develop software that is capable of displaying a 3D volume in 2D slices, and pan through those slices on any of the X, Y, and Z axes
and…
- (stretch goal) is to develop a rudimentary radial averaging program that will at least generate symmetry operators for some angle theta and average a 2D plane of a matrix and report a correlation coefficient. I’m not expecting to get this done yet, but I would be thrilled to at least have a prototype by Jan. 1st.
Also unstated is a way to open and work with these files, as that’s a necessary pre-requisite to doing any of this.