If anyone is looking for a good example of what types of research Folding@Home is contributing to, this one popped up today for me:
In Project 10490, the Chodera lab is studying a small protein called KRAS, which forms a key link in growth signaling and cancer. This gene is something like a molecular switch with a timer. When it is bound to a molecule called GDP, it is off, and does not signal that the cell should grow. However, other proteins can cause it to swap its GDP for a GTP, turning KRAS on. In the on state, it signals that the cell should grow and divide. Normally, after some time, KRAS, with the aid of some partners, will chemically convert its GTP to GDP and return to its inactive state.
In many cancers, this protein becomes mutated, and cannot return to its off state. The result? The cells continue to divide without limit. What’s worse, cancers with this protein mutated tend to have much poorer prognoses. As a result, scientists have been trying to target this protein for decades. So what’s the problem?
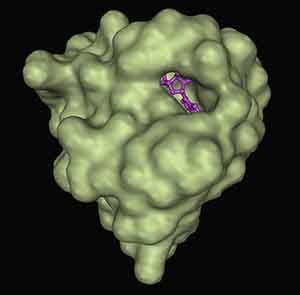
As you can see in the picture above (3GFT in the PDB, for the curious), the only “reasonable’ site for a small-molecule drug to bind KRAS, represented as a green surface, is where the GDP or GTP binds, represented here as purple sticks. However, the affinity that KRAS has for the GDP/GTP is so high that it is considered virtually impossible to outcompete. All is not lost though— proteins move, and in the course of their fluctuations, occasionally expose sites where a molecule could bind. These are invisible on most experimental techniques (like the one used for the previous picture, x-ray crystallography), but may become visible in long molecular simulations. These so-called cryptic binding sites (which have been discovered for other therapeutically interesting proteins like Beta-lactamase using Folding@Home) offer a potential to “drug the undruggable,” and create novel therapeutics for previously intractable diseases.
If anyone is interested in an elaboration, here's what's essentially going on: Basically, what this project is trying to figure out is a way to "shut off" the KRAS protein. That GTP molecule that activates the KRAS protein, which allows the cells to divide is supposed to be hydrolyzed to a GDP after some time, like a natural timer. (GTP and GDP are similar to ATP and ADP, the energy units of the cell.) So the challenge for drugs then becomes, how do we get rid of those GTP that are bound to the KRAS proteins, so that they stop causing the cells to divide?
The traditional way to do this is by inhibition. Basically you find something that the protein can bind to that prevents it from binding to the substrate of matter (in this case, GTP.) As mentioned above, there's really nowhere for anything to bind but the binding site for GTP. However, this is a nefarious case of the protein being too attracted to GTP. Normally, what pharmacists would do is try to figure out where the substrate (GTP) is binding, and create some drug with an even higher affinity to that spot, so that it blocks out the GTP.
This is exemplified in enzyme (or protein) kinetics. For instance, looking at this chart: 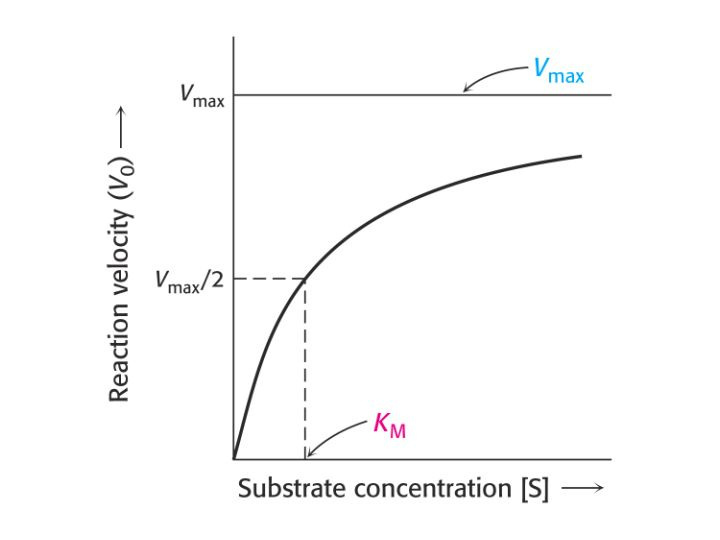
What is depicted here is the enzyme (KRAS protein in our case) to substrate (GTP) reaction. Now it's important to remember, the GTP is supposed to be hydrolyzed to a GDP after a while, but for some reason, that's not occurring. So because that's not occurring, the cell keeps dividing. This chart is nonspecific, but it shows us how these reactions typically occur. The higher the substrate concentration, the faster the faster the reaction will occur. Now this isn't a strict enzyme-substrate reaction, but rather a protein-substrate reaction, because it's not forming a product, but this chart is still good for showing affinity.
So the question then is how do we make something that slows that reaction way down? The best way to slow the reaction down is to prevent the substrate from binding to the protein. The traditional way to accomplish this is via competitive inhibition.
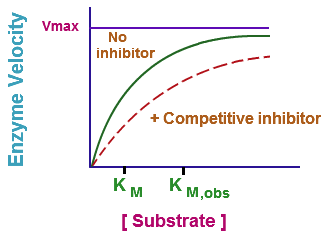
So what's happening in this chart is that some other substrate has been introduced to the protein, and that substrate has a higher affinity for the protein than the original substrate (GTP in our case.) So it's out-competing the GTP. Now, notice how the lines don't just end. The cell can compensate for this with more substrate. So while it might work to inhibit the enzyme, eventually as substrate concentration builds up, it will stop working. This method does not reduce the maximum velocity of the reaction, but rather just slows it until the substrate concentration builds up to a level enough to out-compete the drug, and then it's all systems go once again.
The big issue here is that the concentration and affinity of GTP is too high in the cell to do this. Nothing that researches have placed in that spot on the protein has been able to outcompete the GTP. So when this type of inhibition doesn't work, researchers typically will try noncompetitive inhibition.
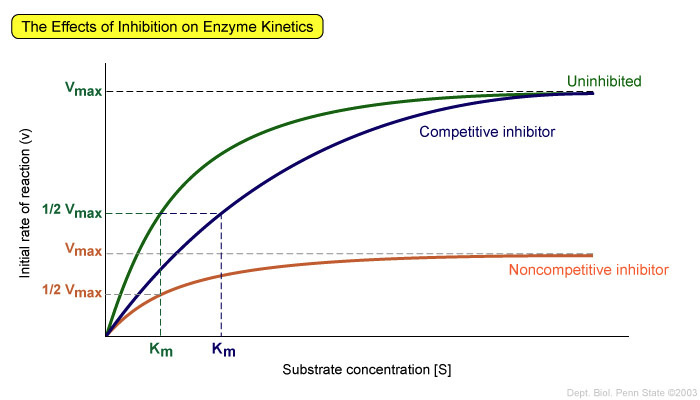
Notice how the line marked "noncompetitive inhibition" is almost a straight line. It barely rises at all, and then it comes to a dead stop. This is because unlike with competitive inhibition, noncompetitive actually reduces the maximum velocity of the reaction, meaning that increased substrate concentration doesn't compensate for the inhibitor. This is because the noncompetitive inhibitor doesn't bind to the substrate binding site in the protein. Therefore it's not competing with the GTP. Instead, it binds to a different site on the protein, and alters the shape of the protein, partially destroying or obscuring the substrate binding site, and isn't vulnerable to being displaced by the substrate because it's not blocking it directly.
The issue noted above, however, is that there is no alternative binding site that has been found on this protein. At least not in its bound state. Many proteins are examined via methods like -xray crystallography. This method of protein visualization is not suitable for examining how the protein interacts with things, or what functions it is performing. It's difficult to observe proteins while they're actually doing work. The hope with this Folding@Home work unit is that we will simulate the protein's movement and work, and that while the protein is moving or working, we can identify a binding site that might otherwise not be there, to place a noncompetitive inhibitor in, and prevent the protein from doing work, and bringing about the unbridled proliferation of new cells in individuals with cancer.